* Stergachis AB, et al. Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science. 2020 의 후속 연구입니다.
본 논문에서 연구진은 Fiber-seq이라는 Long-Read Chromatin Fiber Sequencing 기술을 활용하여 초파리(Drosophila)에서 RNA 폴리머레이스의 원래 크로마틴 환경을 단일 분자 수준(Single-Molecule Precision)에서 시각화했습니다.
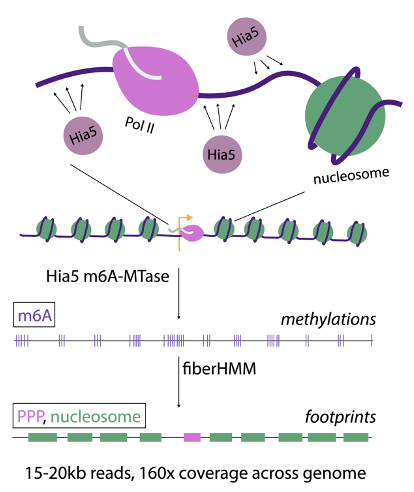
▲ 그림 1. Fiber-seq 기술 개요
Fiber-seq은 m6A-MTase를 사용해 유전체에서 노출된 염기를 표시(메틸화)하고, 이를 PacBio 시퀀싱으로 읽어내는 방식을 의미합니다 (그림 1).
이를 통해 크로마틴 섬유의 물리적 접근 가능성을 단일 분자 수준에서 지도화하여, 크로마틴의 구조와 RNA Polymerase 및 관련 Transcription Machinery의 위치를 연구할 수 있게 합니다.
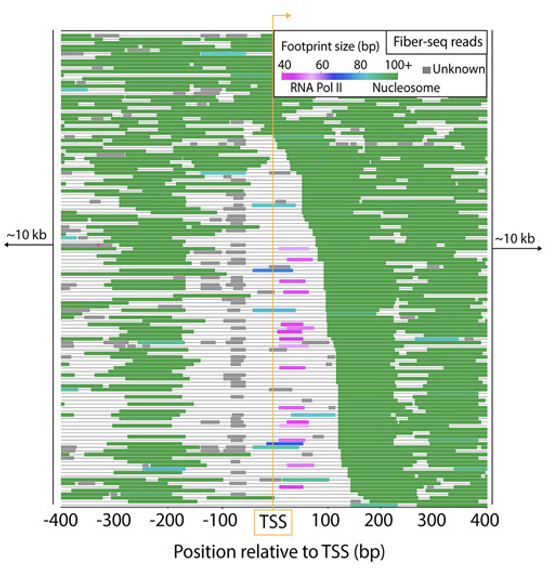
▲ 그림 2. Fiber-seq Reads 및 Footprint
Fiber-seq Reads 데이터는 Footprint를 색상으로 구분하여 특정 크로마틴 구조 또는 단백질-염기 상호작용 영역을 나타냅니다 (그림 2).
PPP (Pink): 전사 개시 전 RNA Polymerase가 Pausing 상태인 영역
PIC (Blue): RNA Polymerase II와 전사 관련 단백질들이 초기 복합체를 형성한 영역
Nucleosome (Green): DNA가 히스톤 단백질에 감겨 뉴클레오솜을 형성한 영역
Unknown (Gray): 구조나 단백질 결합 정보가 불명확한 영역
No Footprint (Black Line): 메틸화나 단백질 결합이 없는, 열린 상태의 DNA 영역
이 색상 구분은 크로마틴 구조와 전사 관련 단백질의 위치를 시각화하여, 크로마틴과 전사 간의 상호작용을 분석하는 데 도움을 줍니다. PPP와 PIC는 전사 개시 상태를, 뉴클레오솜 영역은 크로마틴 압축 상태를, No Footprint 영역은 열린 DNA를 확인할 수 있게 합니다. 결과적으로, Fiber-seq은 크로마틴 구조와 전사 활동을 Long Read Sequencing을 활용해 고해상도로 이해하는 데 유용한 방법입니다.
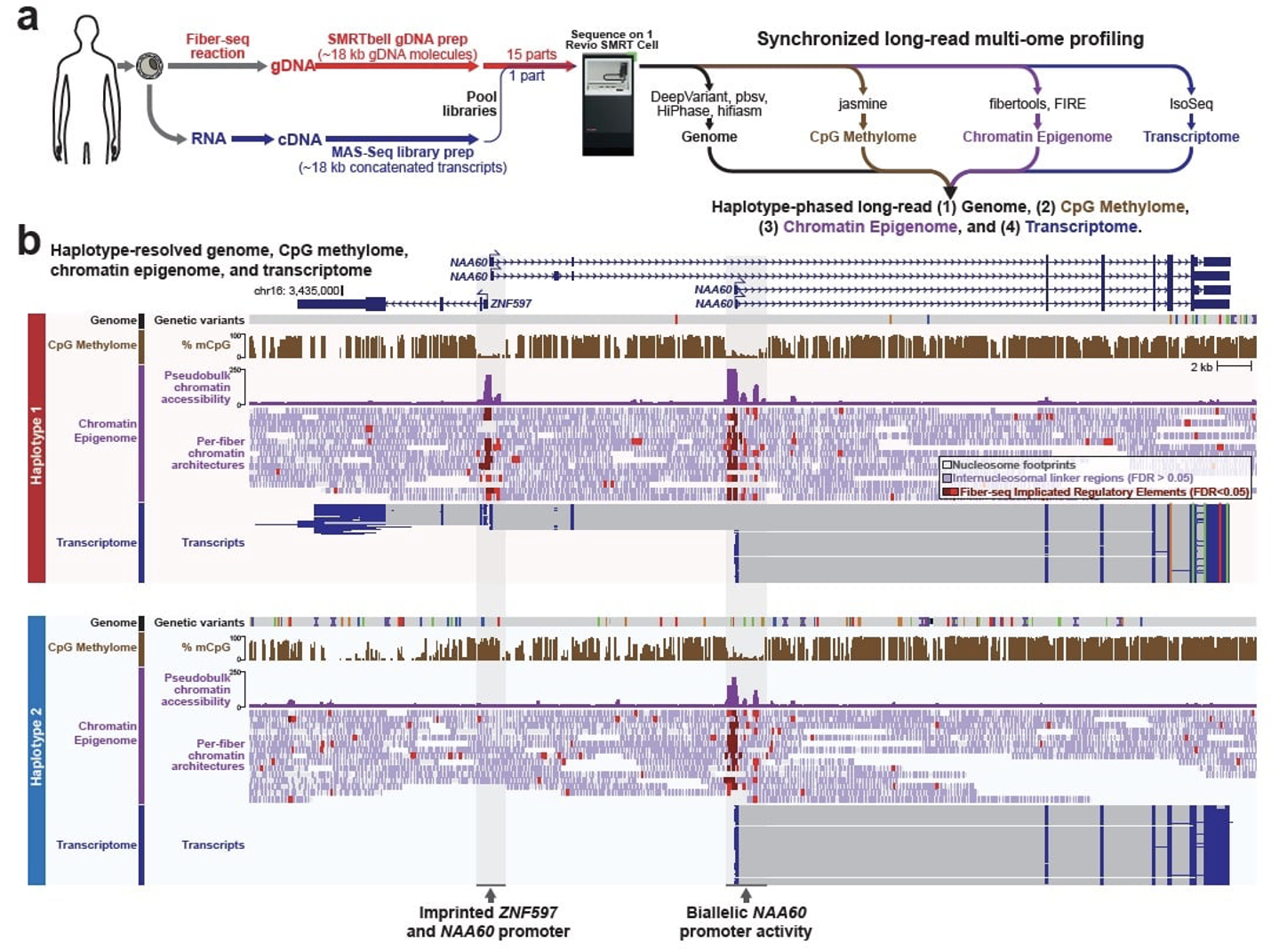
▲ 그림 3. Fiber-seq Reads 및 Footprint의 활용 예시
출처 : Vollger MR, et al. Synchronized long-read genome, methylome, epigenome, and transcriptome for resolving a Mendelian condition. bioRxiv [Preprint]. 2023 Sep
최근에는 이 Fiber-seq과 MAS-seq을 활용한 Multiome 분석 워크플로우가 등장했습니다. (그림 3a)
Fiber-seq: 유전체 DNA를 추출하고 메틸화 정보를 포함한 크로마틴 구조를 분석합니다.
MAS-seq: RNA를 추출해 cDNA를 생성, 전사체 정보를 분석합니다.
이 방법은 유전체(Genome), CpG 메틸롬(Methylome), 크로마틴 에피유전체(Epigenome), 전사체(Transcriptome) 정보를 단일 시퀀싱 실험으로 통합적으로 분석할 수 있는 혁신적 접근법을 제공합니다 (그림 3b).
특히, Long Read Sequencing 기반의 Fiber-seq 기술은 크로마틴 구조, 메틸화 패턴, 전사 활동 간의 상호작용을 동시에 탐구할 수 있게 하며, 이를 통해 복잡한 구조적 변이와 전사 조절 메커니즘의 상세 분석을 가능하게 합니다. 이러한 기술적 이점으로 인해 Long Read 기반 분석에 대한 수요는 앞으로 더욱 증가할 것으로 전망됩니다.
테라젠바이오는 독자적으로 m6A-MTase 단백질의 클로닝, 정제 및 활성 확인에 성공하였습니다. Fiber-seq을 기획하시는 연구자분들에게 협력 연구 조건으로 제공이 가능하오니, 연구자분들의 많은 관심과 문의를 기다리겠습니다.
추가적으로 궁금하신 사항은 이메일 또는 아래 ‘서비스 문의’ 버튼을 통해 문의해주시면 안내드리겠습니다.