안녕하세요, 테라노트 입니다!
안녕하세요 😊 테라노트 입니다.📖
혹시 scRNA sequencing 분석을 진행하다 Data Sparsity의 한계점을 경험한 적이 있으실까요?
scRNA-seq 분석은 단일 세포 단위에서 각 세포의 독립된 전사체 발현 패턴을 확인할 수 있다는 압도적인 장점을 가지고 있지만, 기술적인 요인에서 발생하는 한계점이 존재합니다. 그 중에서, 대표적으로 가장 논의되는 한계점이 바로 Data Sparsity 문제입니다.
오늘은 이러한 Data Sparsity로 인해 발생하는 분석상의 한계점과 문제 해결 방안에 대해 알아보겠습니다.
지난 테라젠바이오 뉴스레터에 대해 궁금하신 분들은 아래 링크를 통해 확인해주세요!
|
|
|
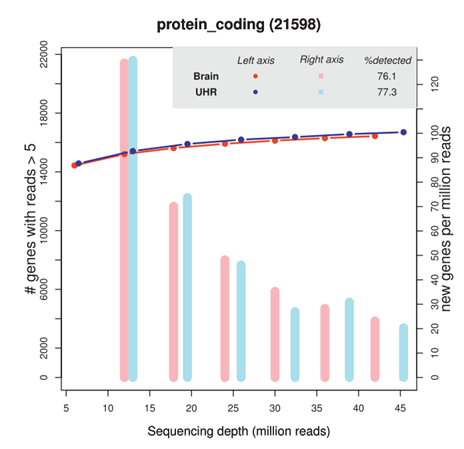
▲그림1. Sequencing depth에 따른 유전자 수 [출처 1] |
|
|
사전 연구들에 따르면, 일반적으로 수행되는 bulk mRNA-seq 분석에서 적절한 sequencing depth는 약 50M reads 수준으로 논의됩니다(그림1).
그러나 scRNA-seq은 설계상 하나의 라이브러리에 5,000~20,000개의 세포에 대한 개별 전사체 데이터가 포함되도록 구성되기 때문에, bulk mRNA-seq과 같은 수준의 depth를 확보하는 것은 사실상 어렵습니다.
실제로 잘 생산된 scRNA-seq 데이터에서도 세포당 확보되는 reads 수는 약 50,000개 수준에 불과하며, 이는 scRNA-seq 분석이 매우 낮은 depth 환경에서 이루어지고 있음을 보여줍니다.
scRNA-seq은 굉장히 많은 세포 수를 대상으로 전사체 발현의 landscape을 확인한다는 강점으로 이를 보완하지만, 이러한 한계점으로 인해 dropout (발현량이 뚜렷하지 않은 유전자가 감지되지 않는 현상) 등의 문제점이 피할 수 없이 발생합니다. |
|
|
scRNA-seq 분석에서 낮은 sequencing depth로 인해 발생하는 dropout 문제를 보완하기 위한 접근 중 하나가 Data Imputation입니다.
scRNA-seq 데이터의 0 표현값(Zero expression)은 크게 두 종류로 나뉩니다.
Imputation의 목적은 이 중 technical zero를 판별하고, 실제로 존재했을 수 있는 진짜 발현값을 복원하는 데 있습니다.
Imputation의 핵심 가정은 "유사한 세포는 유사한 발현 패턴을 가진다”는 점입니다.
대부분의 알고리즘은 다음과 같은 절차를 따릅니다.
-
먼저 감지된 유전자 발현 패턴을 기반으로 세포 간 유사도를 계산합니다.
-
이후 특정 유전자의 결측값(0)을 유사한 세포들의 발현값으로 보완합니다.
예를 들어, A 세포의 유전자 X 발현값이 0이지만, A와 유사한 B·C·D 세포에서 모두 높은 발현을 보인다면, 이 정보를 바탕으로 A 세포의 실제 발현값을 추정하는 방식입니다.
이때 사용되는 방법은 단순 평균부터 복잡한 딥러닝 기반 모델까지 다양하게 적용될 수 있습니다.
Imputation은 적절히 활용될 경우 dropout이라는 기술적 noise를 제거하고, 그에 가려져 있던 실제 발현 패턴을 회복할 수 있어 downstream 분석의 해석력을 높여주는 장점이 있습니다.
하지만 인위적으로 데이터를 생성하는 과정이기 때문에, 새로운 bias를 유발하거나, 세포 간의 미묘한 heterogeneity를 과도하게 smoothing할 위험성이 있어 신중한 사용이 필요합니다. |
|
|
❗ALRA(Adaptively-learned Low-Rank Approximation) |
|
|
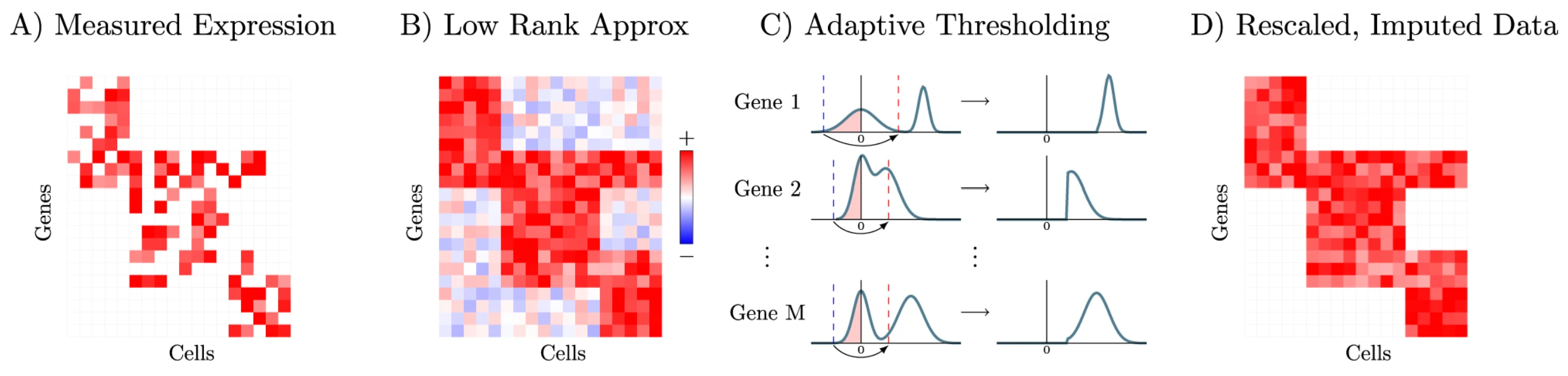
▲그림2. ALRA imputation scheme [출처 2] |
|
|
|
2022년 Nature Communications에 소개된 ALRA(Adaptively-learned Low-Rank Approximation)는 기존 imputation이 갖는 위험성을 효과적으로 완화한 방법론으로 주목받고 있습니다(그림2).
ALRA는 특이값 분해(SVD, Singular Value Decomposition) 방식을 통하여 scRNA-seq 데이터로 부터 데이터 전체의 핵심이 되는 ‘뼈대’를 간추리고, 이 뼈대를 바탕으로 하여 유전자의 발현값을 추정하는 방식을 사용합니다.
기존 imputation 방식들은 유사한 세포 간의 정보를 확산(diffusion)시키는 원리를 따르기 때문에 발현 패턴이 지나치게 평준화되는 over-smoothing이 빈번히 발생할 수 있습니다.
반면 ALRA는 각 유전자의 발현 분포를 기반으로 자동으로 cutoff를 조정하는 adaptive thresholding을 적용합니다. 이를 통해 분해된 특이값(singular values)로부터 벗어나는 유전자들의 발현값을 0 또는 0에 가까운 값으로 처리하여 불필요한 발현 추정을 최소화합니다.
이러한 접근 덕분에 ALRA는 biological zero를 보다 정확하게 보존하고, 세포 집단 간의 미세한 이질성(heterogeneity)을 유지하는 데 있어 기존 방법보다 훨씬 유리한 성능을 제공합니다.
|
|
|
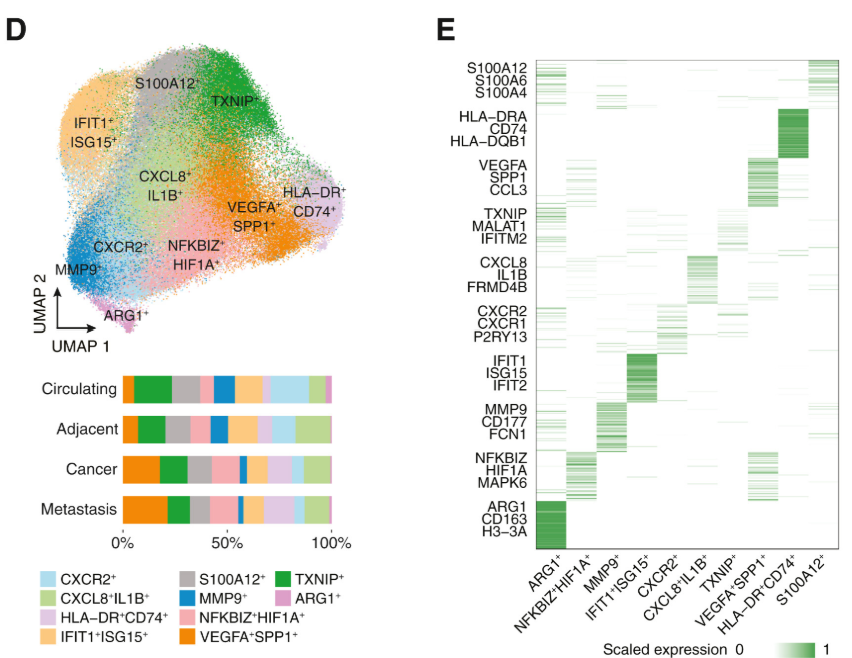
▲그림3. 호중구 proportion, 호중구 subset의 유전자 발현 heatmap [출처 3] |
|
|
ALRA의 뛰어난 성능은 2024년 Cell에 게재된 연구를 통해 더욱 명확하게 확인되었습니다(그림3).
해당 연구는 HLA-DR⁺ 호중구와 T 세포 간의 상호작용을 규명하기 위해 scRNA-seq 분석을 진행했습니다. 그러나 PBMC(Peripheral Blood Mononuclear Cell) 기반 샘플에서 호중구(neutrophil)를 식별하는 일은, 호중구의 낮은 mRNA 함량이라는 생물학적 특성 때문에 본래 매우 도전적인 작업입니다.
연구 저자들은 이 문제를 해결하기 위해 ALRA를 활용하여 데이터를 효과적으로 imputation하였고, 그 결과 기존 방식으로는 탐지하기 어려웠던 호중구를 성공적으로 검출할 수 있었습니다. 뿐만 아니라 ALRA 적용 후 호중구 내부의 여러 subtype을 세부적으로 분류하는 데에도 성공했습니다.
이렇게 정교하게 구분된 세포 타입들은 이후 세포 궤적 분석(trajectory analysis), 세포 간 상호작용 분석(cell–cell communication) 등 다양한 downstream 분석을 수행하는 데 핵심적인 역할을 했습니다. 이러한 분석 결과는 최종적으로 저자들이 주장하는 HLA-DR⁺ 호중구의 기능적 정의와, 관련된 세포 간 상호작용 및 생물학적 결론을 도출하는 데 중요한 기반이 되었습니다.
|
|
|
💡 테라젠바이오의 single cell RNA Sequencing을 경험해보세요!
이처럼 scRNA-seq 분석에서 나타나는 data sparsity 문제는 큰 장애물이 될 수 있습니다. 하지만 ALRA와 같은 전처리(imputation) 기법은 이러한 어려움을 충분히 극복할 수 있음을 보여줍니다.
테라젠바이오는 데이터 품질이 후속 연구의 신뢰도를 결정하는 핵심 요소임을 잘 이해하고 있으며, 이를 위해 자사의 분석 파이프라인 내에 선택 가능한 옵션으로 다양한 imputation 방법론을 적극적으로 도입하고 있습니다.
만약 scRNA-seq 분석을 계획하고 계시지만, 위와 같은 기술적 제약이나 데이터 품질 문제로 고민하고 계시다면, 최적의 분석 결과를 얻을 수 있도록 테라젠바이오가 도움 드리겠습니다!
언제든 편하게 문의해 주세요😊
📄scRNAseq 브로셔 (다운로드)
📄scRNAseq 예시레포트 (다운로드) (다운로드 후 이용가능합니다.)
추가적으로 궁금하신 사항은 이메일 또는 아래 ‘서비스 문의’ 버튼을 통해 문의해주시면 안내드리겠습니다.
그럼 다음 테라노트에서 뵙겠습니다. 😉
테라노트 올림
|
|
|
Follow us!
테라젠바이오 소식을 빠르게 만나보세요. |
|
|
|